Scientists are racing to outsmart cancer
Mohit Kumar Jolly was reading The Emperor of All Maladies: A Biography of Cancer, a popular science book by Siddhartha Mukherjee, when he became impressed by the idea of fighting against cancer. His interest grew exponentially after he started seeing cancer patients every day at the MD Anderson Cancer Research Centre in Houston, USA, where he was pursuing his PhD.
His aspiration to do groundbreaking work in systems biology led him to his current research, in which he uses an integrative approach to decipher how cancer drug resistance develops. “Cancer drug resistance,” says Mohit, now Associate Professor at the Department of Bioengineering, IISc, “is the phenomenon where the same therapy [or drug] that was initially able to control tumour mass is now no longer effective.”
In recent years, researchers like Mohit have taken on the formidable challenge of investigating how cancer cells develop this “superpower” of drug resistance. Scientists have already identified some strategies that cancer cells use, including altering drug-binding sites, ejecting cancer drugs using specialised proteins, or even changing how the human body metabolises a drug.
The human body has long-established mechanisms to keep the spurious growth of tumour cells under check. First in action are innate immune cells, such as natural killer (NK) cells and macrophages, that gobble up the abnormal tumour cells. They also release a barrage of cytokines (chemical messengers) that call other immune cells to the site of action. Dendritic cells are next – they capture the tumour-associated antigens (which act as “signatures” of tumour cells) and present them on their cell surface to alert the adaptive immune system, which comprises cells like T cells and B cells. The adaptive immune system recruits an entire army of cytotoxic killer T cells to fight against the tumour cells.
While all of this sounds organised, the reality is much more chaotic because tumour cells can evolve very quickly. This means that by the time the killer T cell army arrives at the site of action, some of the tumour cells have already changed their identity, and the T cells can’t really recognise them anymore. Think of it as terrorists entering a foreign country being able to generate fake IDs on the fly to fool border security.
By the time the killer T cell army arrives at the site of action, some of the tumour cells have already changed their identity
But what drives such tumour cells to evolve so rapidly?
Sabarinathan Radhakrishnan, Associate Professor at the National Centre for Biological Sciences (NCBS), reveals that genetic mechanisms could be a major factor. “The DNA in our body’s cells undergoes constant damage and repair. [However], sometimes, the DNA replication machinery can introduce mutations, if the damage is left unrepaired, and if it happens in [cancer driver] genes, then those cells take advantage of that and divide rapidly,” he explains.
But mutations are not the only factor. Instead, most tumours use a combination of genetic and non-genetic mechanisms for immune evasion.
“Mutations are not the only or even the major source of drug resistance,” Shaon Chakrabarti, Assistant Professor at NCBS, explains. “[Some of it] has to do with what people call ‘non-genetic’ sources of drug resistance [such as] mRNA and protein levels in a cell, which can vary from cell to cell without any differences in the DNA of these cells.” Think of it as two cells starting with the same raw material (DNA) but depending on their environment (epigenetics) and consequent choices they make at each step of their journey, they end up in different states. These choices are made at the levels of mRNA expression and proteins. Therefore, two cells having the same origin can evolve to show completely different behaviour, such as resistance or sensitivity to drug therapy.
Mohit adds another level of complexity to our understanding of cancer as a dynamic process. He says, “Cancer is not one disease. Cancer is a set of diseases that may manifest very differently in different patients.” This means that we need to start thinking of cancer as “a living system” with myriad evolutionary strategies, he adds.

A complex, dynamic system
Researchers are increasingly realising that tackling the current burden of drug resistance will require integrating cancer biology with multiple fields, including mechanical engineering and physics. For example, since biomolecules like mRNA and proteins are synthesised and degraded at some rate governed by the cell’s internal and external states, physicists like to model these processes as problems of dynamics. A lot of foundational work has already been done in the field of dynamical systems. Thus, physicists and mathematicians, who work on fluctuations and stochasticity (the property of lacking any predictable order) in biological systems, can now test the validity of their theoretical predictions in the cancer drug resistance field.
Mohit’s group is taking such an integrative approach to tackle cancer drug resistance. Their central philosophy is to look at different possible outcomes of a perturbation given to cancer cells. Mohit explains with an analogy of pulling strings in a network. “There are 20,000-30,000 types of molecules in a cell, and they are interacting with each other in complex manners across different length and time scales. When we pull one string out of that network, [assuming] that the string was certainly performing an important role, we might observe beneficial outcomes in terms of changing cellular behaviour,” he says. “In the context of cancer, the expected outcome of pulling that string is that the cell dies. However, what is often not appreciated is that because that same molecule was impacting the behaviour of many others responsible for different functions, other unintended outcomes can also occur.”
He cites the example of tamoxifen, the first targeted drug that came into the market in the 1970s for breast cancer. Tamoxifen can show a dual response: while it effectively kills certain breast cancer sub-populations by repressing estrogen receptor signalling, that same intervention can trigger pathways that make surviving cells more metastatic (easily spreadable to other parts of the body). The critical challenge is quantifying this trade-off – determining how the ratio of cell death versus increased malignancy shifts based on dosage, duration, and a patient’s unique genetic profile.
There is also growing recognition of the need for personalised medicine – shifting from a “one-size-fits-all” approach to focusing instead on an individual’s genetic makeup, lifestyle, and environment, in tailoring treatment regimes.
There is also growing recognition of the need for personalised medicine … in tailoring treatment regimes
To build on this idea, Shaon’s group is working on deciphering how our body’s circadian rhythm (internal clock) plays a crucial role in an individual’s response to chemotherapy. “The same drug given to an individual in the morning versus night can make a very big difference,” he says. His group is also developing methods to extract data on sleep cycles using wearables such as smart rings or smart watches. He adds, “We are developing the maths for a lot of these technologies to personalise the measurement of the circadian time from, say, just a single skin sample. [Using that], can I make measurements and tell you what your body time is?” Their ultimate goal is to use the information about the body’s internal clock to find the right timing to administer chemotherapy and targeted therapy.
Raghuraman Kannan, Professor of Radiology and Biological Engineering at the University of Missouri, has a different approach to tackling cancer therapy resistance. His background in chemistry has helped him develop targeted nano-therapeutics against resistant tumours. He proposes the idea of ‘nanoparticles-as-a-platform’ in which the precursor of a small RNA molecule called siRNA – pro-siRNA – can be attached to a nanoparticle carrier. The pro-siRNA sequence can be customised to silence the genes responsible for an individual’s drug resistance. “The suitcase is going to be the same, but the pro-siRNA inside will be changed in such a way that it can work for personalised medicine,” explains Raghuraman.

at NCBS, preparing cellular samples for RNA measurements (Photo: Adrian Alva)
Despite advancements in producing such nanoparticles, a major bottleneck remains – delivering the drug in a targeted manner to tumour cells. There have been some developments in this area, Raghuraman says, but no unique formula is available yet.
Targeted therapies
There’s a reason why chemotherapy and radiation therapy have remained popular strategies for so long, Sabarinathan points out.
“These [therapies] give more stress to the cell. Given that cancer cells have already lost some of the DNA repair pathways, they can’t tolerate this stress and might undergo cell death. Normal cells – although they will receive some level of damage – because of their proficient DNA repair mechanisms, they would fix the damage,” he explains.
But these therapies alone don’t suffice, as they are quite generic and agnostic to whether a cancer cell or a normal cell receives the stress. Therefore, scientists have started using a targeted approach to specifically attack cancer cells. Sabarinathan adds, “For example, a protein might have a mutation which could change its conformation. Now, we can provide a drug that could directly bind to that altered protein or degrade that protein so that it can stop the function of that mutated protein.”

This approach can help overcome drug resistance in cases where certain proteins are responsible. For instance, a gain-of-function mutation in a protein called EGFR (epidermal growth factor receptor) – which promotes cell growth and division – or a loss-of-function mutation in BRCA1 (Breast cancer type 1 susceptibility protein) – which is involved in DNA repair – can promote tumour growth by altering different pathways of cell survival, growth, and repair. Targeting these mutated proteins specifically could slow down cancer growth.
But there’s a catch. Because cancer cells have poor DNA repair, the few cancer cells that survive treatment will rapidly mutate and grow exponentially. And since DNA codes for mRNA, which is then translated into proteins, these mutated cancer cells can produce more protein variants, enabling them to survive and proliferate. For scientists to develop a clinically approved drug, it takes 10 to 15 years; for cancer cells to make a different protein, sometimes 10-15 days are sufficient.
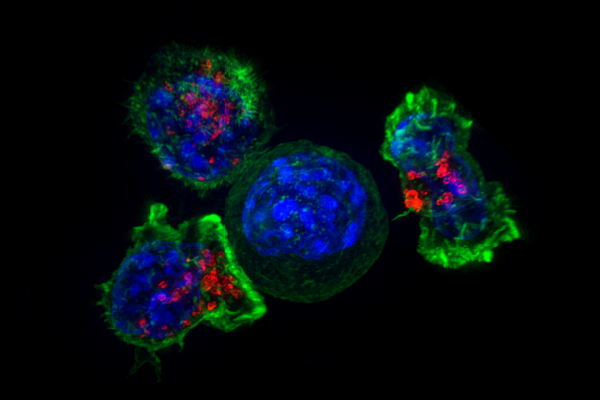
Another promising route that many scientists are pursuing is boosting an individual’s immune system to clear out most of the tumour cells. This is called immunotherapy. Immune cells are capable of rapidly making combinations of different antibodies, with minimal side effects. They are nature’s mini-labs that can make effective and targeted therapies against cancer cells without harming the host.
Another promising route that many scientists are pursuing is boosting an individual’s immune system to clear out most of the tumour cells
The most popular immunotherapy techniques are the use of molecules called checkpoint inhibitors and CAR T-cell (chimeric antigen receptor T-cell) therapy. Some well-known checkpoint inhibitors like PD-1 inhibitors and CTLA-4 inhibitors have shown promise in treating certain cancer types like lung cancer, malignant melanoma (skin cancer), and renal cell carcinoma (kidney cancer). These inhibitors are proteins found on the surface of T cells that help regulate the immune system’s response to cancer. However, the incidence of immune-related side effects, such as fatal hypersensitivity and inflammation, along with exorbitant costs, has remained a barrier for wider adoption of this strategy.
CAR T-cell therapy, on the other hand, modifies a patient’s T-cells to better target cancer cells. The US Food and Drug Administration (FDA) has approved its use for treating leukaemia and lymphoma. However, the limited success of CAR T-cell therapy is often because of the lack of specific targets on solid tumours, which are quite heterogeneous. In addition, the immunosuppressive environment of solid tumours can hinder T-cell function and proliferation.
Despite these challenges, scientists like Raghuraman are optimistic. “Now, immunotherapy works only for 20-30% of the patients, but these 30% of the patients have a very solid response. So, if we put all our energy into making it work for the [remaining] 70-80%, we will have a significant impact on the field.”
Addressing cancer drug resistance will require more than better drugs alone – it will need integrating insights from genetics, systems biology, physics, engineering, and personalised medicine. “We are still not there as a community to be able to cater to all the different challenges that are being faced [by patients] in the clinic,” Mohit says. “So that’s the motivation that keeps [me going].”
Anjaney J Pandey is currently a research scientist at the National Centre for Biological Sciences, Bangalore, and a former science writing intern at the Office of Communications, IISc. He graduated from IISc in 2025 with a BS-MS degree in biology.
(Edited by Ranjini Raghunath)