Researchers, doctors and communities work together to manage a neglected disease

The first time Nagendra Kumar P started experiencing extreme pain was when he was a toddler.
“He had severe headaches,” says his father, B Pandegowda. “We showed him to small hospitals nearby. He would become better for a few days, but the headaches and joint pain kept coming back.”
Pandegowda is a community health worker at the Vivekananda Tribal Health Centre (VTHC) in the Biligirirangana Swamy Temple Tiger Reserve (better known as BR Hills) in Karnataka’s Chamarajanagar district. Like Pandegowda, most people in BR Hills are from the Soliga tribe. In 2006, when Nagendra was around nine years old, he was taken to a hospital in Mysore, where a doctor made an entry in a ruled notebook covered with a jacket made of brown paper, the kind a school child would use. It listed Nagendra’s symptoms and case history, scrawled in the doctor’s handwriting: “Headache – 4 days. Pain abd. – 4 days. Multiple jt. pains – 4 days. Fever – 3 days.” It was the start of a record of a child’s suffering.
The doctor in Mysore diagnosed Nagendra with sickle cell anaemia, a condition that in hindsight, Pandegowda believes had probably affected others in his family too. Pandegowda had a younger brother who used to have frequent pain and was bedridden for months at a stretch; at times he needed someone to carry him to answer nature’s call. The family took the brother to several hospitals, brought him traditional medicine, and prayed to their gods, Pandegowda says, but his brother didn’t survive. Pandegowda’s mother also lived through extreme pain, and in 2012, in her 70s, was so sick that she couldn’t move – her body had curled up into a ball. Doctors in BR Hills had referred her to hospitals in Mysore and Bangalore, but the family couldn’t afford the travel or treatment, and going to Mysore around 90 km away seemed terrifying. “We didn’t have the courage or the money,” says Pandegowda.
In 2006, a month or so after Nagendra’s diagnosis at the Mysore hospital, he fell ill again. His family began the usual rounds of local hospitals and traditional healers. Some people told them that Nagendra would get better as he got older. But even after he started Pre-University College, the headaches and joint pain continued, says Pandegowda. “We were beginning to lose hope … what did we do to deserve this karma?”
The same year that Nagendra was diagnosed, APJ Abdul Kalam, the former President of India, visited BR Hills. He met Prashanth N Srinivas, a doctor and public health researcher who was the medical officer at VTHC at the time, and asked him what diseases were prevalent in the area. Prashanth told the former President about the problem of sickle cell anaemia, to which Kalam replied, “Don’t worry, doctor. Molecular biology, in a couple of years, will solve it.”
Prashanth, who leads the health equity cluster at the Institute of Public Health, adds, “In 2022, the only small tablet we have [as a remedy], hydroxyurea, is still not securely supplied through the government channels. And molecular biology is far away from solving the problem.”
* * *
Although the first official cases of sickle cell anaemia were reported in the USA only in 1910, the genetic mutation that gave rise to the disease is much older. Scientists have traced the disease’s origin to the Sahara desert about 7,300 years ago. In a region as much devastated then as it is now by endemic malaria, the disease may have begun when a mutation turned a child’s red blood cells more rigid and sickle-like, making them a hostile environment for the malaria parasite, and therefore protecting the child from severe disease. This mutation then may have offered protection to generations of Africans from the most severe forms of malaria.
The flip side, however, was that anyone who inherited the genetic mutation from both parents ended up at higher risk for a debilitating condition that came to be known as sickle cell anaemia, also called sickle cell disorder or sickle cell disease (SCD). Today this disease is seen in people whose ancestors come from sub-Saharan Africa, Saudi Arabia, the Mediterranean, and some parts of India.
People with SCD have an unusual form of haemoglobin, the iron-containing protein in our red blood cells that carries oxygen from the lungs to tissues and organs, and brings carbon dioxide from these sites back to the lungs for disposal. While haemoglobin molecules are typically globular in shape, the sickle haemoglobin (HbS) molecules stick to each other, forming long rigid structures inside a cell, giving them the characteristic sickle shape. People who inherit the gene mutation from one parent are said to have a condition called sickle cell trait (SCT), while those with genes inherited from both parents will have a 25% chance of being born with SCD. In communities that practise endogamy – marrying within their own community – the likelihood of having two parents who are carriers is higher.
People with SCD have an unusual form of haemoglobin, the iron-containing protein in our red blood cells that carries oxygen from the lungs to tissues and organs
The reason why SCD is of concern from an individual as well as a public health perspective is that its symptoms can be severe. Under certain conditions, when blood flows through tiny blood vessels in the body called capillaries, the sickle-shaped red blood cells end up clumping together and blocking the blood vessels. Blocked blood vessels cut off oxygen supply to vital organs, leading to organ damage. In many patients, particularly children, these blocks occur in the spleen, causing the kind of severe abdominal pain that Nagendra frequently experienced. Many children with a severe form of the disease do not survive beyond the age of five. Sometimes, patients feel such extreme pain that they have said they feel like killing themselves, says Deepa Bhat, a doctor, genetic counsellor and Associate Professor at JSS Medical College in Mysore.
In addition to extreme pain crises, which can severely impact the sufferer’s ability to lead a normal life, SCD can cause the death of tissues like bone or joints, recurring infections, strokes, severe anaemia, acute chest syndrome, and meningitis. People across the country experience different symptoms based on their geographical location and the community to which they belong (the prevalence of the sickle cell trait varies from 1% to 40% among tribal groups in India). Because symptoms such as jaundice, severe joint pain or recurring infections are not unique to SCD, doctors sometimes erroneously write them off as arthritis or put routine infections down to lack of hygiene, according to Deepa.
This is not just an Indian problem. Even though SCD is one of the most prevalent genetic disorders in the world, according to the WHO, most countries do not have national-level control or systematic screening programmes, or even basic facilities to treat patients. In the USA, where SCD largely affects BIPOC (Black, Indigenous, or People of Colour) communities, mostly Black Americans, systematic marginalisation and a resulting lack of trust in the country’s healthcare system has meant that many have slipped through the cracks. Gene therapy or bone marrow transplants – the only possible cures – are extremely expensive, can only be performed on some patients, are relatively new, and at the moment unavailable in many countries including India. The long-term effects of gene therapy for SCD are also unknown. There is no cure, therefore, for many SCD patients – the only way is to manage the symptoms.
But even managing symptoms is not easy. SCD is an overlooked disease in India, largely because of social and institutional neglect. Until recently, it was considered a disease that only affects tribal populations because of its high prevalence among them. According to the Ministry of Tribal Affairs (MoTA), one in 86 children in Scheduled Tribe communities is born with SCD. Accurate numbers are hard to come by as data on tribal health remains insufficient. Poor access to doctors also means that just maintaining general health is hard for these vulnerable populations. Due to a severe lack of awareness about the disease, even among doctors, patients are rarely advised about even simple strategies to manage the symptoms, such as avoiding dehydration, overexertion or high altitudes, all of which can trigger a pain crisis.
Like some other diseases in India, the actual number of people suffering from SCD is likely to be much higher than official figures – researchers have repeatedly flagged the glaring absence of data. And the problem lies in the way screening for the disease is done currently. For one, the tests have many practical and logistical challenges. Secondly, screening is largely only in tribal communities. But doctors and medical researchers have pointed out that they have seen patients from non-tribal populations with extremely severe presentations of disease – a phenomenon that is relatively less known and accounted for in public health programmes.
“What we are seeing,” says Deepa, “is only the tip of the iceberg.”
* * *
Expanding screening might be a social problem, but developing tests that are more effective at picking up the disease falls squarely within a scientist’s domain. And some like Sai Siva Gorthi have been working to address this issue.
An Associate Professor and Gore Subraya Bhat Chair in Digital Health at the Department of Instrumentation and Applied Physics, IISc, Sai first heard about the challenges related to screening for SCD a few years ago when he was contacted by Nisanth Nambison, an associate professor and nodal officer for a tribal sickle cell project at the Government Homoeopathic Medical College and Hospital at Bhopal. By then, Sai and his team had already been working on developing point-of-care diagnostic devices, and he had founded a startup called ShanMukha Innovations to take these devices to the market.
When Sai met Nisanth, the latter explained that he had just started working on a project funded by MoTA to test the effectiveness of homoeopathic treatments for sickle cell anaemia in particularly vulnerable tribal groups – the Baiga and Bharia tribes – in Madhya Pradesh. For the project, he first needed to collect blood samples from thousands of people in the Dindori, Mandla and Chhindwara districts, and identify those who had the disease using the solubility test.
The solubility test, the most common screening test for the disease today, is also the least expensive. A doctor or nurse takes a tiny volume of blood from a needle prick and dissolves it in a test tube containing sodium hydrosulphite, which specifically precipitates the abnormal form of haemoglobin (HbS) found in sickle cell carriers, making the solution turbid. Turbidity is an indication that the person has sickle cell abnormality, but the doctor can’t decide that a patient is a carrier or has the disease from just looking at these murky samples. The next step is to send the patients’ blood samples to a lab that has the facility to carry out High Performance Liquid Chromatography (HPLC) – the current standard for diagnosis – which identifies the presence of HbS more accurately.
“Both in terms of sensitivity and specificity, the solubility test is not great, and it doesn’t provide information about whether the patient is positive for sickle cell trait or disease,” says Sai. The HPLC test is more expensive and requires a greater amount of blood (about 3-5 ml), which can scare off some patients, because it needs to be drawn from the vein by an experienced technician or nurse. And blood samples collected at remote locations in tribal areas need to be shipped to labs or district hospitals which can sometimes be hundreds of kilometres away. “After we painstakingly collect the blood and take it to the district hospital, the person doing the HPLC will not be available, or will be on leave, or the machine will be broken down. Our effort of getting that 3 ml of blood from the patient would have gone to waste,” explains Nisanth.
Blood samples collected at remote locations in tribal areas need to be shipped to labs or district hospitals which can sometimes be hundreds of kilometres away
Such delays can cost lives. Nisanth recalls an instance when his team had taken a blood sample from a child living in a remote location for further testing to a faraway district hospital. But rains played havoc in the following weeks and the child’s report also became misplaced due to a clerical error. By the time they recovered the report – which showed her as positive for the disease – and returned to the same village, a month had passed and the child had died.
Given these challenges, Nisanth asked Sai if he could come up with a device that would immediately and accurately spell out the test result at the point of blood collection itself, using just a tiny volume of blood. Spurred by Nisanth’s appeal, and using the blood samples he shipped to IISc, Sai’s team at ShanMukha Innovations began developing a diagnostic test. During their explorations they made an important discovery. When blood samples from both normal and sickle cell patients are stripped of oxygen using a buffer solution, the abnormal haemoglobin molecules undergo unique conformational changes. This causes the diseased samples to absorb light differently compared to healthy samples, a change that can easily be detected using a simple, portable spectrophotometer. Although some scientists had noticed earlier that normal and diseased blood show differences in light transmission, no one else had reported specific, measurable differences in light absorption properties before, explains Sai. “That was the starting point.”

Excited by this discovery, the team developed two versions of the new technology they named as HPOS (High Performance Optical Spectroscopy) which uses a combination of a small hand-held, portable digital reader and reagent kit to treat the tiny drop of blood; SickleFind, which would help screen for individuals who might have either sickle cell disease or trait, and SickleCert, which can help distinguish between sickle cell disease and sickle cell trait.
Once they had the process nailed down, they had to work on getting approval for marketing and selling the kit. They were keen on getting approval first from Indian regulators – a longer and more arduous process than getting a USA FDA or EU approval, which most other competitors were doing. “We wanted to make sure we launch the product in India first,” says Arun Balasubramanian, Director of ShanMukha Innovations. They first obtained a test licence, which would allow them to carry out a clinical study in a lab accredited by the National Accreditation Board for Testing and Calibration Laboratories (NABL). The next step was to get a manufacturing licence from the Central Drugs Standards and Control Organisation (CDSCO).
The new technology uses a combination of a small hand-held, portable digital reader and reagent kit
But just as their efforts were gathering steam, COVID-19 hit. “All the field workers who were collecting blood samples were redirected to COVID-19 efforts. There was nothing happening for sickle cell, or malaria or any other disease,” says Arun. “We could not get samples, and there was no technical support available to perform any kind of study.” They finally managed to get about 120 blood samples for the clinical study, test their kit and submit the data to CDSCO in November 2021. But the CDSCO rejected the grant of manufacturing licence in the first round after a lengthy audit, and asked them to carry out additional accelerated studies to strengthen the case for their claim that the product had a six-month shelf-life. In November 2022, after they resubmitted their data to CDSCO, their sickle cell kits were finally approved, clearing the way for them to market and sell the product. They have also been advised by the Indian Council of Medical Research (ICMR) to carry out a multi-centre study with samples from different regions of the country. “They would like us to test samples with all variants,” Arun says.

Sai Siva Gorthi, Rajesh Srinivasan, Prateek Katare, Bhaskar Varanasi (Photo courtesy: Rajesh Srinivasan)
However, acceptance of such products by doctors and healthcare authorities, and incorporation into their screening programmes – “mindshare”, as Arun puts it – will take time and effort. Tanya Seshadri, Chief Medical Officer at VTHC and Programme Head at the Centre for Training, Research and Innovation in Tribal Health (CTRITH) in Karnataka, says that a device like this would certainly be useful for the purpose of screening for sickle cell trait, but she would rather use the current gold standard test to make a definitive diagnosis of something as serious as SCD. “It’s labelling someone for life without being absolutely certain first. We are not yet at a stage where staff at the periphery – at the primary health centre (PHC) level – are equipped to handle confirmatory testing, patient counselling and follow-up,” she says, adding that she would prefer to wait until a product like this is proven to have a low rate of error.
While Deepa believes it is important to have confirmation at the point of care, she also feels that SickleCert has some way to go to eliminate the likelihood of false negatives, and that the process could be made simpler. Currently, she says, it involves many steps – a technician mixing the blood sample with a reagent, waiting 20 minutes, inserting it into a spectrophotometer, and interpreting the results – and this requires training and careful handling. “At the PHC, realistically, you cannot control these things,” she says. She currently prefers the two user-friendly rapid test kits that operate like pregnancy test kits – SickleScan, which is used extensively in Africa, and Hemotype SC, which is made in Canada and now distributed in India. “We took feedback even from ASHAs and ANMs [Accredited Social Health Activists and Auxiliary Nurse and Midwives] – they like these because it involves just a finger prick and gives instant results.” But both Deepa and Tanya are supportive of the concept of an indigenously developed low-cost, simple testing kit, given the extent of the problem in India.
* * *
“At first I thought, ‘I’m gonna come in here and fix this,’” says Tanya, recounting her initial naivety about SCD as a doctor hoping to intervene in a public health problem. However, good intentions, she points out, aren’t enough to grapple with the complicated reality of providing healthcare to vulnerable populations.
In 2018, when Nagendra was studying for a degree in commerce, the pain was becoming unbearable. His family began another round of visits to doctors in the hope of finding a solution. Nagendra spent time in Malavalli, Maddur and Gundlupet meeting doctors and trying every treatment possible, including Ayurvedic ones. At various points, he was given protein powder, calcium tablets, and folic acid tablets – none of which served as a solution.
At the moment, patients with pain crises are given painkillers and blood transfusions to reduce the complications caused by the blocking of vessels and increase the oxygen-carrying capacity of the patient’s blood. In order to reduce the frequency of such crises, patients are prescribed hydroxyurea, a cancer drug that increases the level of foetal haemoglobin, which is thought to lessen the severity of the disease. Hydroxyurea, which costs around Rs 15 per capsule, has to be taken every day, for life. A new treatment is currently being developed by researchers in the USA – a drug to stimulate production of an enzyme called pyruvate kinase in red blood cells, which might help alleviate pain crises.
Good intentions, Tanya points out, aren’t enough to grapple with the complicated reality of providing healthcare to vulnerable populations
Beyond just treating the symptoms, scientists are looking at the possibility of curing SCD. In 1949, the biochemist Linus Pauling and his collaborators identified anomalies in haemoglobin as being the cause for SCD, showing for the first time that an abnormal protein could be linked to a disease, and that genes determined the structure of proteins. New developments in the field of gene editing, particularly the development of CRISPR-Cas9 in 2012, have accelerated the possibility of gene therapy as a cure – a patient’s genes can be modified to ‘correct’ the sickle cell mutation or increase the levels of foetal haemoglobin, also known as gamma haemoglobin. Until a baby is born, it depends on gamma, which is suppressed a few months after birth and then beta haemoglobin takes over. Editing the stem cells of a patient with SCD to avoid the suppression of foetal haemoglobin might give them a fighting chance against the disease.
Vasanth Thamodaran, Senior Scientist at the Tata Institute for Genetics and Society (TIGS) in Bangalore, explains that foetal haemoglobin levels of 20-40% can protect against severe disease. In order to achieve these levels, haematopoietic stem cells (HSCs, the precursors of all blood cells) are ‘mobilised’ from the bone marrow of a patient (brought into circulating blood), extracted, and edited – using CRISPR-Cas9 to target the repressor binding region in the cells, which limits the suppression of foetal haemoglobin – before being reintroduced into the patient’s body. “Clinical trials of this method have already been done in the USA, and they are transfusion-independent, so we know the concept works,” he says. Researchers at Christian Medical College (CMC), Vellore have also shown that it can be done in pre-clinical studies using mouse models, he adds.
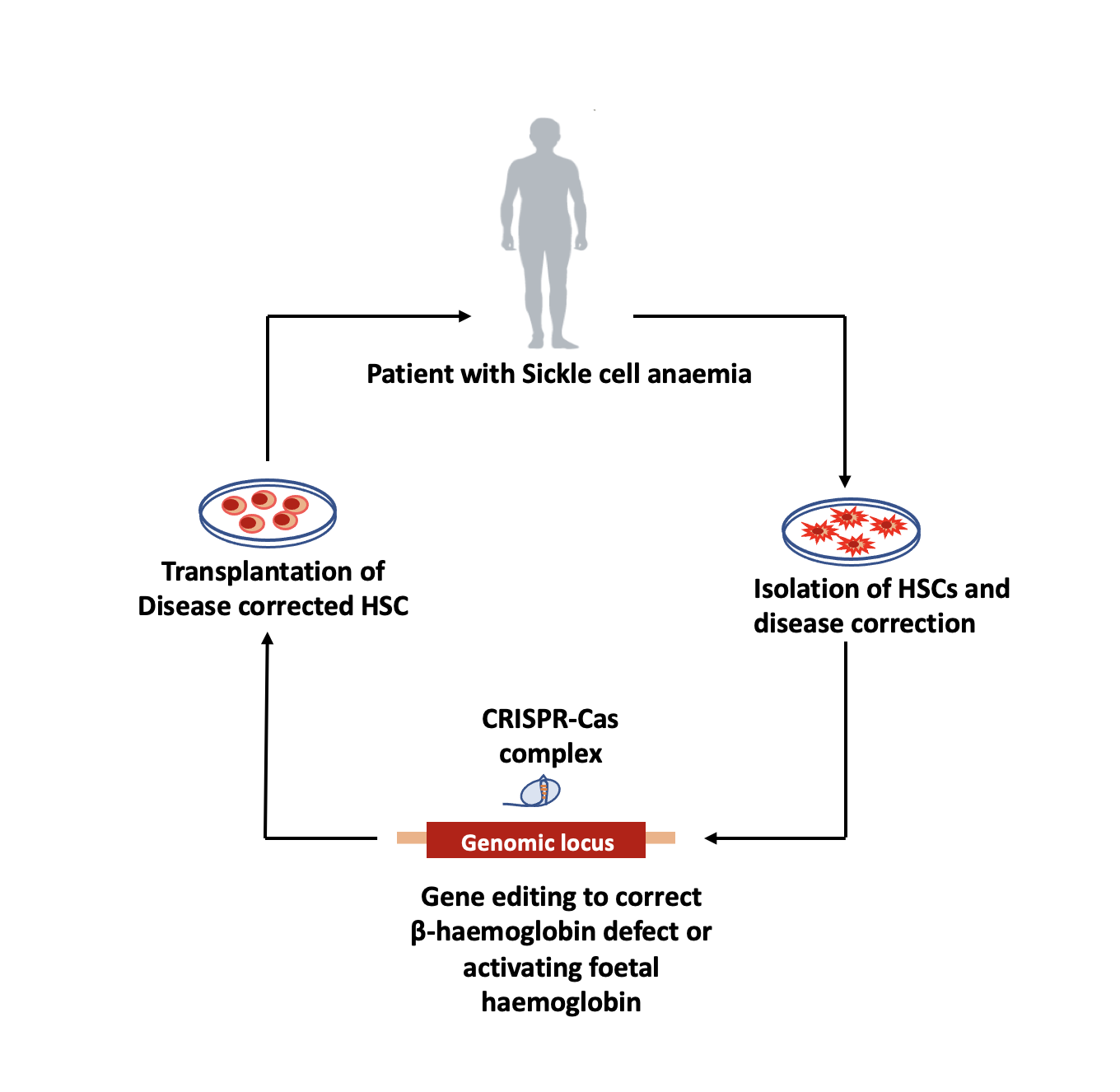
“What we are trying to do [at TIGS] is to reduce the cost of the procedure, which is estimated to be around 900,000 to 2.1 million USD.” Many of the components involved in gene editing currently have to be imported, which drives up the cost. The TIGS team’s focus is on the culture medium in which the isolated stem cells are placed during the process of editing, which requires five ‘growth factors’ – protein molecules that they use to prevent stem cells from differentiating into other kinds of cells, such as red blood cells, which will render the treatment ineffective. “Our goal is to make those growth factors in-house at a lower cost,” Vasanth says, adding that they are also focusing on using smaller CRISPR-Cas9 systems to bring down costs.
“But technology is only one part of medicine,” writes physician Dhruv Khullar in the New Yorker about gene therapies for SCD. “[M]any of the people who need them the most are on the fringes of a medical system that has marginalized them. Sickle-cell disease traces the deep, long-standing inequities of American society. Defeating it will require confronting them.”
Part of the reason that many Soligas like Nagendra receive inadequate treatment is an enormous fear and hesitation to approach doctors until they reach the point of desperation – like it happened with his grandmother – because they do not trust that they will be treated with dignity. Even for those who overcome their fear and approach doctors, the likelihood of getting the right diagnosis and advice isn’t guaranteed. Some doctors at the PHC level or district hospital level – the first point of contact for people who live in remote rural areas – seem to be unaware of SCD and how to effectively treat or manage it, according to Deepa and others.
C Madegowda, a Soliga researcher and community leader, says that when his nephew suffered severe joint pain at the age of 10, he took him to a government hospital in Mysore. Madegowda, who has a PhD in social work focused on his community, is a postdoctoral fellow at the Ashoka Trust for Research in Ecology and the Environment (ATREE), and is the current secretary of the community organisation Zilla Budkattu Girijana Abhivruddhi Sangha, says, “They didn’t connect his problems to sickle cell anaemia. Because I knew about the disease and the fact that it affects Adivasis, I asked the doctors to test him for sickle cell anaemia. They didn’t have a facility to test for it, so they took a blood sample and sent it to Bangalore. Then he received a diagnosis, and was able to get treatment.”
The Soligas are just one community among the 50 or so tribes in Karnataka, which make up nearly 7% (numbering around 42.5 lakh people, according to the last Census in 2011) of the state’s population. And Soliga patients are only some of the many people affected by sickle cell disease that Deepa sees on a regular basis. Since 2019, Deepa has been a Principal Investigator for the National Task Force under which six states have worked together to develop a comprehensive model applicable at the level of PHCs, to screen for and manage SCD.

Research by Deepa’s team was largely conducted in HD Kote and other tribal belts in the Mysore district that are home to a range of people from tribal backgrounds, including Jenu Kurubas, Betta Kurubas, Yeravas, Paniyas, and others. The team learned that the hospitals where communities could avail of care were usually far from their settlements, and the expectation that patients would travel these long distances was unrealistic. Screening in Karnataka, if at all it was carried out, only targeted those between six and 21 years old instead of all ages, including newborns. Decisions on the healthcare model were made by the authorities without seeking the participation of the affected communities. People who did participate in the screening did not hear back about their results. Even after being diagnosed, patients were not receiving a regular and free supply of hydroxyurea tablets to manage their pain, and many of them struggled to get a disability card, which would qualify them for pension, free blood transfusions and other benefits. The work they’d begun continues under CTRITH, where, among other areas of focus, they aim to build a registry of people with SCD who can be targeted for treatment and follow-ups.
Deepa believes that screening is futile without efforts to create awareness about the disease and provide follow-up treatment after diagnosis. Her team identified local leaders trusted by their respective communities (such as Ratnamma, who has a PhD in sociology focused on the Soliga community), and worked with them to spread the word about the disease and how to manage it, as well as to identify people in crisis so that they can receive timely treatment. “We created awareness, screened people, and supplied hydroxyurea,” says Deepa. “We also helped build a referral system: we trained medical officers to identify mild, moderate and severe symptoms, and the courses of action necessary in each case, including sending people with severe cases requiring painkillers that cannot be administered at PHCs, to hospital by ambulance.”
Deepa believes that screening is futile without efforts to create awareness about the disease and provide follow-up treatment after diagnosis
Still, overcoming decades of mistrust in the public health system can be hard for the communities. Deepa says, “People would ask us, ‘Why are you taking our blood? Are you drinking it? Selling it?’ They have even chased us away.”
* * *
In 1986, when doctors Abhay Bang and Rani Bang moved to the district of Gadchiroli in Maharashtra, they diagnosed a 10-year-old girl with suspected heart disease as having SCD – the first identified case of SCD in the entire district. They then began a district sample survey and found 1 lakh carriers of the sickle cell gene and nearly 6,000 people with SCD. As a result of their much-lauded efforts, a centre for tribal medical research was set up – but in Pune, not in Gadchiroli, as they had requested. Recounting the matter, Abhay, who received a Padma Shri in 2018, writes in Ideas for India: “Disappointed, we approached the tribal leaders in the villages and requested them to put some pressure on the government to bring the centre to Gadchiroli. Their response took us unawares: ‘Doctor, this is your disease, not ours,’ they said. ‘Did we ever tell you that we need help for this?’”

The tendency to see tribal people as data points – or “guinea pigs” for scientific curiosity as Abhay writes – has left many of them wary of sickle cell programmes conducted by researchers and the government. Madegowda says with an air of fatigue, “Doctors have a project, they have funding for it, they need to show progress reports. They present papers at conferences and receive recognition. But the community receives no treatment. Everybody comes to take blood samples from our community. But nobody wants to share the results.” NGOs in the area have projects for one or two years, and then when the funding dries up, the projects are rolled back, he says. “Ultimately, who is facing the problem? We are facing the problem,” he says, highlighting the need for members of the community to volunteer to spread awareness about the disease and its treatment as well as the difficulties of doing so. For tribal community leaders like him, advocating for better healthcare is yet another battle they have to fight for their people.
Madegowda, who is a carrier of the sickle cell gene, adds that if it had turned out that he did have the disease, he wouldn’t have wanted to know. “Telling me I have the disease will create automatic stress, it will create stigma. If someone tells me, ‘Oh, you have sickle cell anaemia,’ I would feel guilty, like it is my fault,” he says. Deepa says she has seen people hide the fact that they have children with SCD or fail to disclose their carrier status in matrimonial alliances because of the stigma. She adds that counselling people to not marry other carriers can have adverse socio-cultural impacts, and doesn’t believe in doing so herself. “There is life beyond carrier status,” she emphasises.

Madamma, a Soliga community leader from Muthagada Gadde Podu in BR Hills, and a member of the same community organisation as Madegowda, believes she shouldn’t allow the disease to hold her back. Having suffered chronic pain since childhood and experienced pain so severe during her pregnancies that she feared she wouldn’t survive, she tries to ensure the disease doesn’t affect her daily life. “I don’t think much about having a disease. I should scare this disease a bit, I feel.”
“I don’t think much about having a disease. I should scare this disease a bit, I feel”
For others without Madamma’s grit, doctors can play a big role in providing positive reassurance, according to Madegowda. “Doctors have to … reassure them that their condition can be treated or managed and that they may be able to lead a normal life, and build their confidence rather than tell them there is no hope for them,” he says. “That is why we prefer to go to our oracles and traditional healers, or to temples. They give us hope.”
Hope finally came to Nagendra when he was started on hydroxyurea by Deepa in January 2022. After two decades and nearly 30 different doctors, it was a turning point – the pain had lessened and Nagendra was finally experiencing some relief from the disease that had plagued his childhood and adolescence. “Only because I’m taking the tablet now, the pain is under control. Otherwise, the suffering is intense,” he says. In the past, Nagendra was never diligent about taking daily medications like folic acid tablets. “Now, he takes the medicine regularly on his own,” says his father Pandegowda.
It’s not just the patients who are persevering. For scientists like Sai and Arun, developing a diagnostic device for a “neglected” disease like sickle cell anaemia is a hard sell. “Not just in diagnostics, in pharma too, people only make drugs for which they know there is a big market. There are a lot of neglected conditions, or small pockets of challenges or issues which nobody cares about,” says Arun. But what keeps them going, he says, is the promise such technologies offer to save lives and the commitment they see among the clinicians and researchers they have interacted with so far.
“We researchers are dedicated,” says Deepa. “But we cannot do much without government policy, which needs more streamlining.” She has seen funds for the SCD programmes bounce around in various departments or return because of technicalities, and a lack of focus as multiple sickle cell groups operate simultaneously without adequate coordination. But she hasn’t given up. “We are here to work.”
Prashanth also emphasises the role of the government in fighting the disease alongside scientists. “Innovation that IISc produces may have meaning in patients’ lives, but without state support, it is tough.” The number of people who might need a product like a low-cost screening test for SCD may be high, he says, but they are also less likely to be able to pay for it. “In the interests of equity and justice, someone has to pay, and it has to be the state.”
With input from Samira Agnihotri