From simple combinations of lenses, microscopes have evolved into complex tools giving us unparalleled insight into cellular secrets

Mondal Lab, IAP, IISc. PhD research scholars (from left: Prakash Joshi,
Neptune Baro, Aravinth S, Jigmi Basumatary) working on the system (Photo courtesy: Partha P Mondal)
January 1665 saw the publication of Robert Hooke’s Micrographia. The beautiful illustrations of fleas, flies and other insects in it gave humanity an insight into the sheer beauty of the hitherto unobserved world of small creatures. The book sold more copies than any other scientific work published before it, earning it the sobriquet of “the first scientific bestseller”.
Hooke had used a novel tool to look at these creatures: a microscope. But his most enduring legacy was something else he saw through its lens – tiny, repetitive structures in cork, resembling the small rooms which served as the homes of Christian monks. He named these structures cells.
The main themes of Robert Hooke’s work have remained with biologists ever since. To use new and better techniques, and to get clearer and sharper images. And of course, to look at cells.
Towards ever sharper imaging
Since Hooke’s time, the field of microscopy has become instrumental in understanding biological phenomena. And within this field, the technique of fluorescence microscopy stands out. This involves tagging proteins or subcellular components of interest with a “fluorophore” (usually a short protein) which will emit light when excited with light of a particular wavelength. When photons from the light source hit the electrons in the fluorophore, they gain energy and release it in the form of light at another, longer wavelength. This emitted light can then be captured and used to locate the object to which the fluorophore is bound.
“Biologists typically seek to understand two major challenges: Self-assembly of molecules, when they come together to create larger and larger structures, and the biological functions these structures are involved in or give rise to,” explains Deepak Nair, Associate Professor at the Centre for Neuroscience (CNS), IISc. “Microscopy seeks to understand the locations where these functions manifest.”
“This has been one of the greatest revolutions of our time,” adds Rahul Roy, Associate Professor at the Department of Chemical Engineering, IISc. “Nowadays, you cannot publish a paper in biology without imaging the cells or animals you are studying under a microscope.”
Fluorescence microscopy can be used on both stained and fixed cells as well as live cells. One can use it to understand how a cell was at a particular point in time, or to understand the dynamics of certain proteins as they move about inside a living cell.
“Nowadays, you cannot publish a paper in biology without imaging the cells or animals you are studying under a microscope”
Until recently, confocal microscopy was the cutting edge of fluorescence microscopy. Confocal microscopes use lasers to illuminate the sample and gather light from just the focal plane (the depth into the sample where the microscope’s lens is focused), and not from other components behind or in front. This leads to an increase in the ‘signal-to-noise ratio.’
Unfortunately, a typical confocal microscope is limited by the ‘diffraction limit’ of light. The resolving power of a microscope is inversely proportional to the wavelength of light being emitted. If fluorophores are closer than a certain distance from each other – the limit is typically 250-300 nm – the microscope is unable to resolve them as separate objects.
Can you super-res that?
But the limits of confocal microscopy have not deterred biologists from wanting greater resolution. A powerful new technique that aims to break through this limit is superresolution microscopy. In confocal microscopy, all the fluorophores of interest get excited at once. “But in superresolution microscopy, you can excite them stochastically, meaning only some of them can be active and fluoresce at a time. This allows for resolution of distances up to 20-30 nm,” explains Partha Mondal, Professor at the Department of Instrumentation and Applied Physics, IISc.
In fact, these fluorophores blink, and using careful imaging techniques, one can figure out how many photons are being emitted by the molecule as well as the location of the blinks. Plugging this data into a mathematical formula enables one to get the centroid and size (enabling precision in localisation) of the blinking molecule. “At this stage,” Partha remarks, “you are playing at the level of single molecules.”
Partha’s lab has worked on developing multiple superresolution techniques. Some examples are POSSIBLE (Probabilistic Optically-Selective Single Molecule Imaging Based Localisation Encoded) superresolution microscopy, which allows for resolving objects less than 10 nm in size, and SMILE (Simultaneous Multiplane Imaging-based Localisation Encoded) microscopy which allows for scanning the entire volume of a cell.
Another method for increasing resolution is a technique called DNA-PAINT, used by Mahipal Ganji, Assistant Professor at the Department of Biochemistry, IISc. “Our lab looks at the organisation of DNA in the cell,” he explains. “The molecule is two metres long and compressed into a small volume. We look at events happening within this volume using DNA-PAINT.”
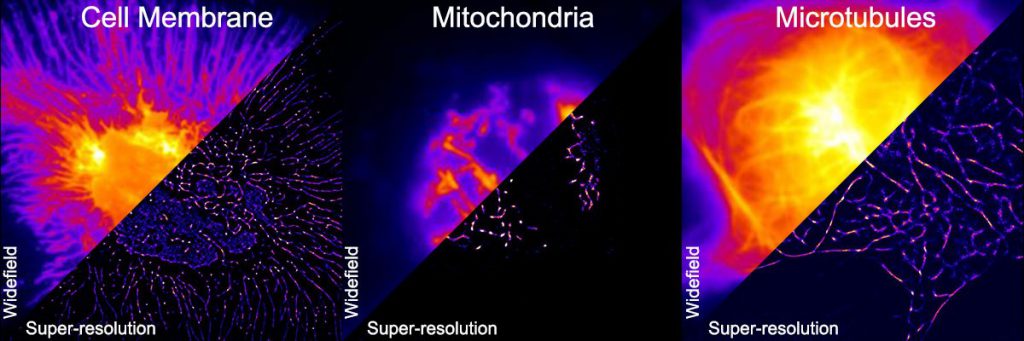
(Image courtesy: Deepak Nair)
DNA-PAINT works on the principle that two complementary DNA strands – constructed synthetically – will bind with each other. This binding is short-lived at room temperature, causing them to bind and unbind randomly. Mahipal and his team typically attach one DNA molecule to the protein they wish to study and later add a fluorophore-conjugated DNA molecule to the solution in which the cell is being imaged. Every time the fluorophore-conjugated DNA binds to its partner, a small flash of light appears as the attached fluorophore emits fluorescence. “This results in blinking,” continues Mahipal, “which is essential to a lot of superresolution microscopy techniques.”
One advantage of this technique is that the fluorophores do not “photobleach”. In most types of fluorescence microscopy, the repeated activation of the same fluorophore makes it no longer fluoresce after a point. In DNA-PAINT, since the binding of fluorophore-conjugated DNA is completely random – the same molecule is not necessarily involved each time – the chances of photobleaching are low.
Unfortunately, DNA-PAINT imaging times tend to be very long. “It is not yet practical to use this technique on live cells,” concedes Mahipal. “But the advantage is that once we’re done with one protein, we can wash the fluorophores off and add a different strand of DNA which binds to another protein attached with corresponding partner DNA.” Since the proteins are being imaged sequentially, one can use the same fluorophore for each protein. “We can add an imager for a protein, image it, then wash it off, and repeat this for as many proteins as we have prepared the cell for.”
This is in stark contrast to most other techniques in which researchers must attach a different fluorophore emitting light at a different wavelength to each protein of interest. “You can only image around 2-3 different proteins in the conventional way, because that is the number of unique fluorophores you can practically image together,” says Mahipal. “In contrast, DNA-PAINT can potentially image hundreds of proteins sequentially.”
Mahipal uses a kind of microscope called a total internal reflection fluorescence (TIRF) microscope to perform DNA-PAINT. It works on the principle that light which hits glass at a particular angle does not get refracted at all, but gets completely reflected. The point at which light hits the glass produces an ‘evanescent wave’, an electromagnetic wave that remains localised at the point it is produced. This evanescent wave then goes on to excite fluorophores at the surface of the cell.
The same kind of microscope is also used by Rahul. “We employ TIRF or a light-sheet based method called HILO (Highly Inclined Illumination) to track single molecules,” he explains. These methods allow a high signal-to-noise ratio during imaging of single molecules, and therefore enable capturing their motion in real-time.
Rahul’s lab uses it to examine the way in which molecules assemble into complexes by themselves. Another technique he uses is single molecule Fluorescence Resonance Energy Transfer (smFRET) to study protein shapes and their interactions with each other. This technique involves a donor and an acceptor fluorophore. When the two fluorophores are very close to each other, the donor transfers energy to the acceptor, causing the acceptor to fluoresce. “This technique allows us to measure extremely short distances of around 1-10 nm, making this a very precise way of measuring interactions. So if two molecules come together to form a complex, this technique will report that.”
“You can gain an unprecedented level of insight into the function of a cell through such techniques,” explains Deepak. His lab works on understanding the organisation of neural synapses in the brain at extremely fine resolutions. They have built a modular microscope which allows them to perform different operations by merely switching out some parts. It is capable of being used for superresolution microscopy as well as other advanced techniques such as single-molecule tracking, optogenetics, and Fluorescence Recovery After Photobleaching (FRAP).
There are various other methods of getting to superresolution as well. “The past decade or so was the time of confocal microscopy,” says Partha. “But the future is all superresolution and light sheet microscopy techniques.”
Sheets of light
Despite being powerful techniques, confocal microscopy and superresolution microscopy have a major flaw: they tend to scan a plane point by point. This is often too slow to follow events happening in a live cell.
Light-sheet microscopy tries to solve this problem by illuminating an entire plane of light at once. “The illumination can be a rod or a lattice, depending on how the microscope is constructed,” explains Rahul. “Either way, you image much more than one point at a time.”
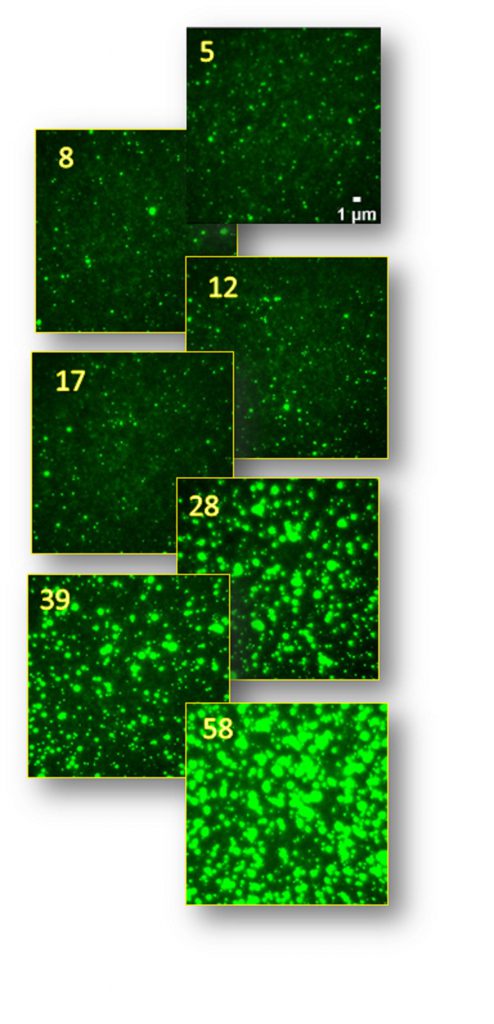
Partha’s lab has worked extensively on light-sheet microscopy techniques. “The real advantage of light-sheets,” he states, “is that you get amazing temporal resolution. Since a plane isn’t being scanned point by point, you can get very good images at very short time intervals.”
“Light-sheet microscopy also has a great advantage in areas where you wish to image a wide field of view at resolutions near the diffraction limit,” he adds. “One can work with large samples such as zebrafish and
C. elegans.”
In 2013, Partha’s lab demonstrated an integrated light-sheet imaging and flow-based enquiry (iLIFE) system. “We were able to integrate light sheets into microfluidic devices to carry out imaging cytometry,” he says. “The channels in a microfluidic device are only wide enough for a single cell or organism (such as C. elegans) to flow through. The iLIFE system is able to scan them as they flow through, allowing the interrogation of a large population of cells. This is something which has never been done before.”
Photon machine gunning
Some questions, however, require slightly different types of tools to answer. “Our lab is interested in understanding how information is stored in the brain,” says Balaji Jayaprakash, faculty member at the Centre for Neuroscience, IISc.
His lab performs experiments on mice and track neuronal activity in the live brain. The typical tool at hand for such studies is the venerable MRI. “[However], the resolution we require is at an altogether different level,” he explains. “We study synapses, which are of the order of microns. MRIs have a resolution of a few millimetres.”
Which is why they use a technique called two-photon microscopy. “We open a window on the mouse’s skull and image their brains,” he continues. “Instead of using a photon with the exact energy needed to excite a fluorophore, we use two photons with half the energy required for excitation.”
The realisation of this technique in practice was enabled by the development of ultrafast lasers which send a large burst of photons at once, like machine gun fire. This is key to building such microscopes, because the photons have to strike the fluorophore very quickly for the required energy transfer to take place.
The advantage of using low-energy photons is that they have greater penetrating power. For example, brains, when seen under the microscope, appear white, because the cells scatter all incident visible light. But infrared rays, which have lower energy than visible light, can penetrate some distance into a mouse’s brain. This allows for the excitation of fluorophores at a greater depth. “The microscope we have built can image at depths up to 1 mm, and a few more adaptions can reach 1.5 mm,” says Balaji.
Balaji explains that the microscope has enabled them to come up with a new kind of plasticity mechanism for how the brain integrates new memories. “Traditional explanations for memory formation focus either on the generation or the strengthening of synapses. We have been able to see that there is another mode for information storage: spatial rearrangement of synapses,” he says.
Unfortunately, superresolution is not yet possible in multi-photon microscopy. “It is a trade-off,” Balaji explains. “If you want to go deeper, you have to sacrifice some resolution. But people are working on it. It might not be here today, but it will come.”
The path ahead
“The big thing about microscopy is that it is highly accessible,” says Rahul. “Commercially available microscopes are becoming more and more powerful and accessible for researchers. People who work on microscopy also like to make their techniques more user-friendly so as to enable the wider community to benefit.”
Deepak concurs. “Ever since the advent of confocal microscopy, the modularity of microscopes has been increasing. You can swap out one module for another depending on the functions you wish to access. This allows people who are not interested in instrumentation per se to also use extremely cutting-edge technology for their experiments. [Microscopy] has thus become one of the most democratised techniques in everyday research.”
“[Microscopy] has thus become one of the most democratised techniques in everyday research”
At the same time, the cutting edge of microscopy is also accelerating. Researchers are striving to combine light-sheet and superresolution techniques in a single setup. New reporter molecules are being developed that can tell scientists more than just the location of molecules. Tools to “bar-code” and distinguish individual neurons within a brain are also on the horizon. The advent of artificial intelligence (AI) and machine learning (ML) is transforming the way state-of-the-art microscopes acquire, store and evaluate large data sets.
Such developments will eventually allow instrument limitations to stop being an obstacle. “That truly is the dream,” remarks Deepak. “Your work should only be limited by your ambition.”
Savyasachee Jha is a PhD student in the Department of Management Studies and a former science writing intern at the Office of Communications, IISc